The cotton genetic improvement team at Huazhong Agricultural University recently published a research paper on the transcriptional regulatory structure of allotetraploid cotton in Nature Communications.
This research initiated the Cotton Encyclopedia of DNA Elements (ENCODE) project, exploring the dynamic changes in subgenomic three-dimensional structure and epigenetic modifications throughout the cotton lifecycle, elucidating the regulatory mechanisms underlying subgenomic expression asymmetry, and assessing the genetic effects of non-coding elements in fiber quality improvement.
Allotetraploid upland cotton, formed by the doubling of A and D genome diploid ancestors, is a widely cultivated cotton variety. The team has long been studying the evolutionary divergence and coordinated regulatory mechanisms of its subgenomes, and over the past three years, it has brought to light the mechanisms by which subgenomes regulate fiber development and quality, providing theoretical support for the design and breeding of high-quality cotton.
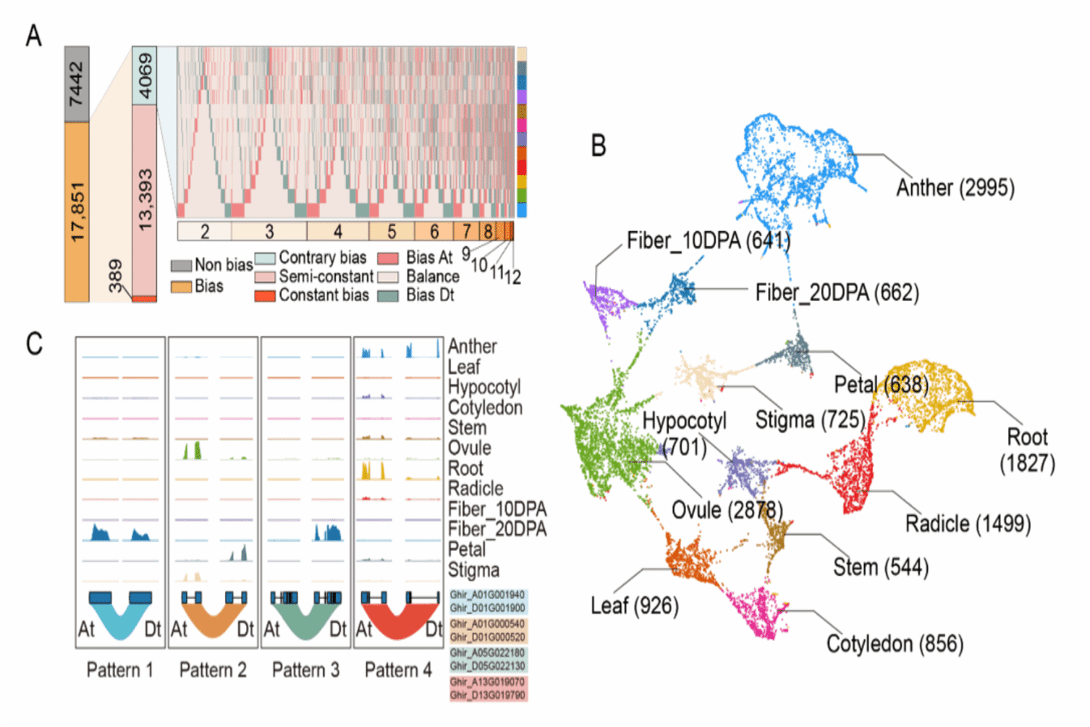
Expression map of homologous gene bias/tissue dominance during whole growth stage of cotton [Photo/news.hzau.edu.cn]
To comprehensively understand the functional genome of cotton, the study conducted an in-depth analysis of the transcriptional bias of subgenomic homologous genes throughout the lifecycle based on chromatin three-dimensional structure and epigenetic genome status.
Approximately 70 percent of subgenomic homologous genes exhibited biased expression, with tissue-specific expression genes showing more pronounced bias, according to the study.
The study also analyzed the multilevel subgenomic structural features across multiple cotton tissues, revealing that the Dt subgenome undergoes more frequent state transitions between tissues and proposing the concept of tissue-specific highly active TADs (TS-TADs). The rearrangement of subgenomic TAD-like domains may be a crucial factor contributing to the expression divergence of subgenomic homologous genes.
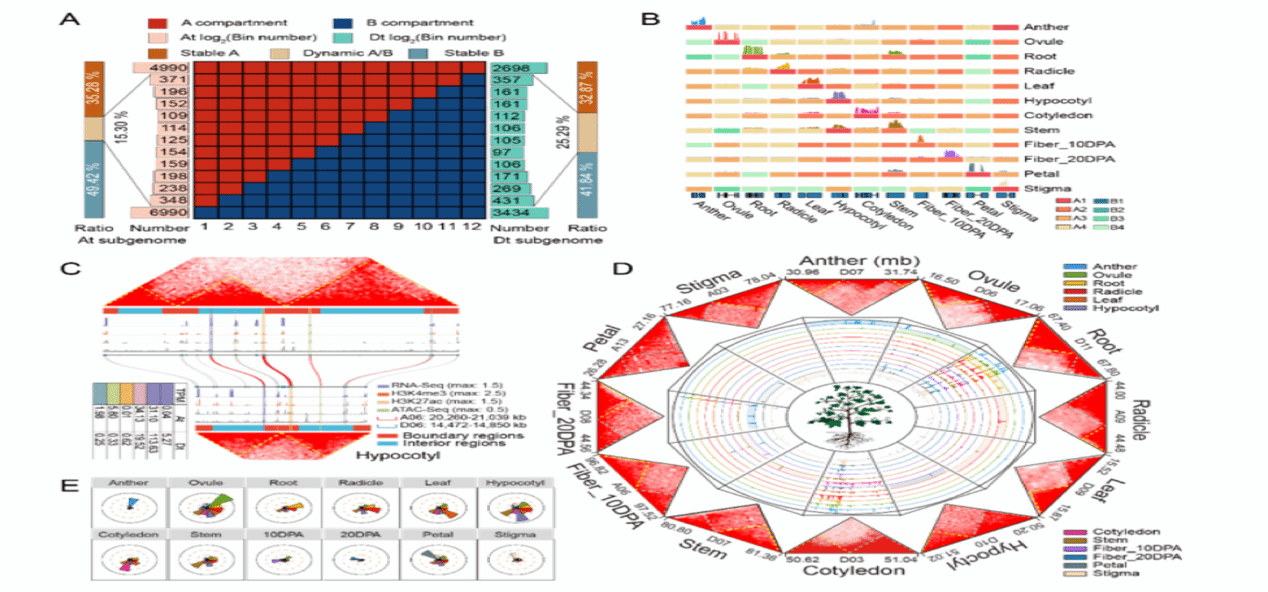
Multilevel three-dimensional spatial structure of cotton subgenome [Photo/news.hzau.edu.cn]
Furthermore, the study developed a method for predicting tissue regulatory elements (PATREs), combining histone modification signals to predict potential functional elements in non-coding regions. The analysis found that subgenomic three-dimensional spatial rearrangement and differences in chromatin epigenetic modification status are key factors leading to inconsistent regulation of homologous genes.
By integrating transcriptome data, the study evaluated the potential of cis-regulatory elements (CREs) in pyramiding breeding for fiber quality improvement, discovering that an increase in the copy number of certain CREs is positively correlated with fiber quality, and that there are underutilized CREs with good breeding potential.
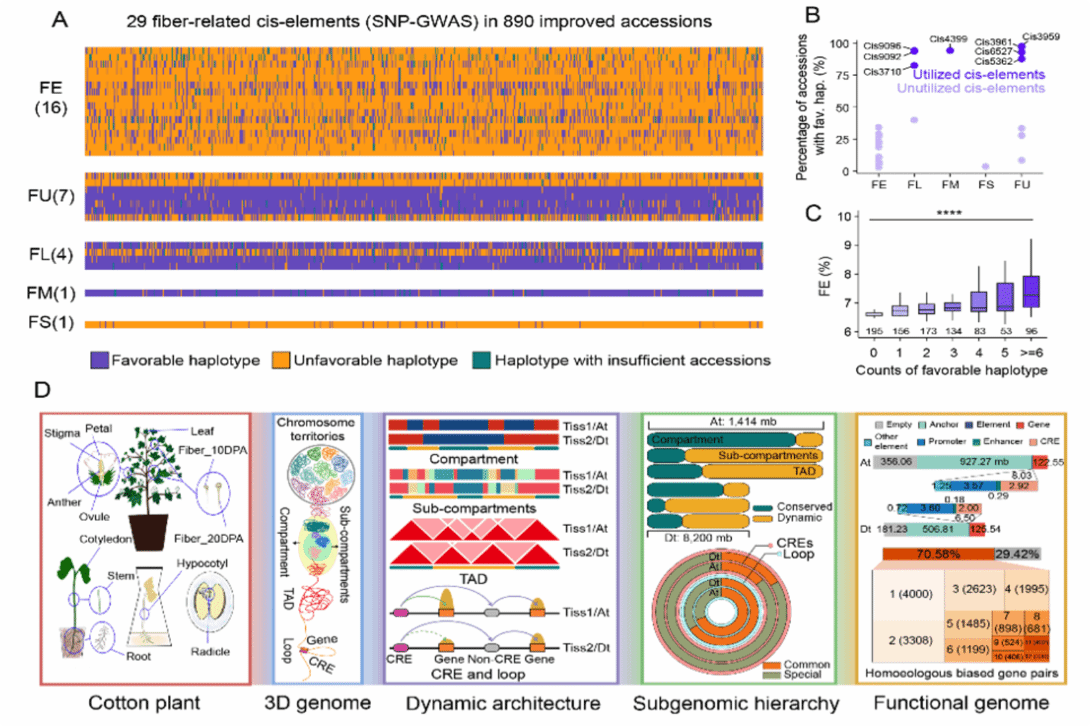
CRE drives cotton fiber quality improvement. [Photo/news.hzau.edu.cn]
Finally, the study established the Cotton ENCODE website, a publicly available database that helps promote the in-depth analysis of the cotton genome and the development of functional genomics.
The authors of the paper include Huang Xianhui, Wang Yuejin, Zhang Sainan, et al., with Professor Wang Maojun as the corresponding author. Academician Zhang Xianlong and Professor Zhu Longfu also participated in the research. The study was supported by funding from institutions such as the National Natural Science Foundation of China.






















